EXAFS calculation
From FEFF
Contents |
Estimate of 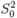
All of the examples in this section yield calculations for the K edge (default). To do calculations for other edges, use the EDGE or HOLE card. These cards will also yield an estimate of from atomic calculations if you set S02 = 0 by the one of three possible ways shown below.
EDGE L3 0.0 HOLE 4 0.0 S02 0.0
The result for S02 is given in ‘chi.dat’ or ‘xmu.dat’ files. is a square of determinant
of overlap integrals for core orbitals calculated with and without core hole. The core-valence separation can be changed by editing subroutine getorb, but it is currently set by default to the most chemically reasonable one.
Configuration averaging over absorbers
In amorphous materials or materials with distortions from regular crystals, the absorbing atoms (with the same number in the periodic table) may have different surroundings. Thus one may want to average the calculation over different types of sites for the same atom or even over all atoms in the ‘feff.inp’ file. This can be accomplished using the CFAVERAGE card. The usefulness of this type of calculation is currently curtailed by the limited functionality of the CFAVERAGE card, which should be used with caution.
Adding self-consistency
Self-consistency is expected to be more important for XANES calculations, but even for EXAFS one may want to have a more reliable determination of Fermi level or to account for the charge transfers in order to do fits with single energy shift E0. Our experience shows that reliable EXAFS phase shifts are best achieved using the SCF card.
SCF 3.8
The above example works for solids or large molecules, but for molecules with less than 30 atoms, calculations can be done faster if you set lfms1 = 1,
SCF 10.0 1
For details see the SCF and FMS cards.
Calculation Strategies and Examples
EXAFS calculations
- SF6 Molecule
- Solids
- Estimate of S20
- Configuration averaging over absorbers
- Adding self-consistency
