Initially the free atom potentials of each atomic type are calculated as if the atoms were isolated in space using a relativistic Dirac-Fock atom code. Scattering potentials are calculated by overlapping the free atom densities within the muffin tin approximation (Mattheiss prescription), and then including the Hedin-Lundqvist/Quinn self energy for excited states. Non overlapping muffin-tin radii are determined automatically from calculated the Norman radii. Automatic overlapping of muffin tin spheres (see the AFOLP card) is done by default, since it typically leads to better results than non overlapping muffin-tin spheres. FEFF8 can also calculate self-consistent potentials by successively calculating the electron density of states, electron density and Fermi level at each stage within a small cluster and then iterating, using the Mattheiss prescription for the initial iteration.
XAFS spectra are referenced to the threshold Fermi level. This quantity is best determined with the self-consistent field procedure (typically to within a fraction of an eV), or (less accurately but faster) can be estimated from the electron gas result at the mean interstitial density within Mattheiss prescription, as in FEFF7. An absolute energy scale is obtained by an atomic calculation of the total energy of the system with and without the core-hole. Atomic configurations and core-hole lifetimes are built in, and mean free paths are determined from the imaginary part of the average interstitial potential, including self-energy and lifetime contributions.
The potential calculations need as input only the atomic number of the atoms, and, for the absorbing atom, the type of the core hole being considered. To do the overlapping of the unique potentials, the neighboring atoms must be identified, either by position (from a list of the cartesian coordinates of each atom) or by explicit overlapping instructions using the OVERLAP card described in Section 2.3.
To save time the code calculates the overlapped atom potential for each unique potential only once, using as a sample geometry for an atom with a a given unique potential index that for the atom that is closest to the absorbing atom. Thus it is essential that the neighborhood of each sample atom be appropriate.
* touching muffin-tins; do not use automatic overlapping AFOLP 1.0
* L1-shell core hole, S02 = 1 EDGE L1 1.0
The HOLE card includes the hole-code index and the amplitude reduction factor ![]() . If the entry for
. If the entry for ![]() is less than 0.1, then
is less than 0.1, then ![]() will be estimated from atomic overlap integrals. Experimental values of
will be estimated from atomic overlap integrals. Experimental values of ![]() are typically between 0.8 and 1.0. The defaults if the HOLE card is omitted are ihole=1 for the K shell and
are typically between 0.8 and 1.0. The defaults if the HOLE card is omitted are ihole=1 for the K shell and ![]() =1. The hole codes are shown in Table 2.2.
=1. The hole codes are shown in Table 2.2.
FEFF is designed to calculate absorption from completely filled shells. You can try to simulate absorption from valence electrons with FEFF, but you may get unreliable results. If you encounter difficulties and need valence shell absorption, please contact the authors.
For ![]() , the core-hole lifetime parameter (
, the core-hole lifetime parameter (![]() ) is not tabulated in FEFF and is set equal to 0.1 eV, since the final state losses are then dominated by the self-energy. Use the EXCHANGE card to make adjustments (
) is not tabulated in FEFF and is set equal to 0.1 eV, since the final state losses are then dominated by the self-energy. Use the EXCHANGE card to make adjustments (![]() ).
).
* K-shell core hole, S02 estimated by overlap integrals HOLE 1 0.0
![]()
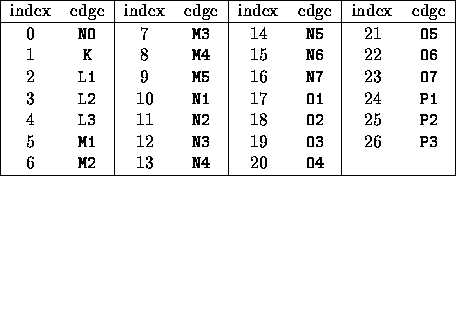
Table 2.2: Available hole codes. The entries in the columns marked edge are written as they are recognized by the EDGE card. Index 0, NO, is the no hole option described in the NOHOLE card.
* ipot Z [tag lmax1 lmax2 xnatph]The required list entries are the unique potential index ipot and the atomic number Z. The tag is at most 6 characters and is used to identify the unique potential in the 'path00.dat' output file. The optional list entries lmax1 and lmax2 are used to limit the angular momentum bases of the self-consistent potentials (XSPH) and full multiple scattering calculations (FMS). If a negative number (e.g.,
 ) is specified for either lmax1 or lmax2, FEFF will automally use a default based upon the atomic number of the species normally lmax(atomic). The last optional entry xnatph can be used to specify the stoichiometric number of each unique potential in the unit cell of a crystalline material. This helps in the calculation of the Fermi level. In the case of an infinite solid,
) is specified for either lmax1 or lmax2, FEFF will automally use a default based upon the atomic number of the species normally lmax(atomic). The last optional entry xnatph can be used to specify the stoichiometric number of each unique potential in the unit cell of a crystalline material. This helps in the calculation of the Fermi level. In the case of an infinite solid, The absorbing atom must be given unique potential index 0. These unique potential indices are simply labels, so the order is not important, except that the absorbing atom is index 0, and you may not have missing indices (ie, if you use index 3, you must also have defined unique potentials 1 and 2).
To save time the code calculates the overlapped atom potential for each unique potential only once, using as a sample geometry the first atom in the atom list with a given unique potential index. Thus it is essential that the neighborhood of that sample atom be representative. Failure to do so may cause the code to generate inaccurate potentials and phase shifts and poor XAS results.
* molecular SF6 Sulfur K edge, lamx1=default, lmax2=3 (spdf basis)
POTENTIALS
* ipot Z tag lmax1 lmax2
0 16 S -1 3 1
1 9 F -1 3 6
The S02 card specifies the amplitude reduction factor ![]() . If the entry for
. If the entry for ![]() is less than 0.1, then the value of
is less than 0.1, then the value of ![]() is estimated from atomic overlap integrals. Experimental values of
is estimated from atomic overlap integrals. Experimental values of ![]() are typically between 0.8 and 1.0.
are typically between 0.8 and 1.0.
* let FEFF calculate S02 S02 0.0
* -10% overlap of muffin tin with unique potential 0 * 10% overlap of muffin tin with unique potential 1 FOLP 0 0.9 FOLP 1 1.1
EXCHANGE ixc vr0 vi0 (ixc0) (Useful)
The EXCHANGE card specifies the energy dependent exchange correlation potential to be used for the fine structure and for the atomic background. ixc is an index specifying the potential model to use for the fine structure and the optional ixc0 is the index of the model to use for the background function. The calculated potential can be corrected by adding a constant shift to the Fermi level given by vr0 and to a pure imaginary ``optical'' potential (i.e., uniform decay) given by vi0. Typical errors in FEFF's self-consistent Fermi level estimate are about 1 eV. (The CORRECTIONS card in Section 2.8 is similar but allows the user to make small changes in vi0 and vr0 after the rest of the calculation is completed, for example in a fitting process.) The Hedin-Lundqvist self-energy is used by default and appears to be the best choice for most applications we have tested in detail. The partially nonlocal model (ixc=5) gives slightly better results in some cases, but has not been tested extensively.
Another useful exchange model is the Dirac-Hara exchange correlation potential with a specified imaginary potential vi0. This may be useful to correct the typical error in non-self-consistent estimates of the Fermi level of about +3 eV and to add final state and instrumental broadening.
Defaults if EXCHANGE card is omitted are: ixc=0 (Hedin-Lundquist), vr0=0.0, vi0=0.0. For XANES, the ground state potential (ixc0=0) is used for the background function and for EXAFS the Hedin-Lundqvist (ixc0=0) is used.
*Hedin-Lundqvist -2eV edge shift and 1eV expt broadening EXCHANGE 0 2. 1. *Dirac-Hara exchange -3 eV edge shift and 5 eV optical potential EXCHANGE 1 3. 5.
This card roughly simulates the effect of complete core-hole screening. It is useful to test the final state rule for calculated XAS, and to compare to other calculations (such as band structure or other codes) that do not use a core hole. The code will use as the final state that specified by the HOLE card for the matrix element calculation -- the NOHOLE card will cause FEFF to calculate potentials and phase shifts as if there is no core hole. For dDOS and ![]() or
or ![]() absorption calculations, for example, NOHOLE often gives better agreement for white line intensities. Conversely NOHOLE tends to give poor XANES intensities for K-shell absorption in insulators such as BN.
absorption calculations, for example, NOHOLE often gives better agreement for white line intensities. Conversely NOHOLE tends to give poor XANES intensities for K-shell absorption in insulators such as BN.
The radial grid used for the potential and phase shift calculation is
![]()
with ![]() by default. The default is sufficient for most cases. However, occasionally there convergence problems in the atomic background at very high energies (the background curves upward) and in the phase shifts for very large atoms. If such convergence problems are encountered we suggest reducing delta to 0.03 or even 0.01. This will solve these problems at the cost of longer computation times (the time is proportional to
by default. The default is sufficient for most cases. However, occasionally there convergence problems in the atomic background at very high energies (the background curves upward) and in the phase shifts for very large atoms. If such convergence problems are encountered we suggest reducing delta to 0.03 or even 0.01. This will solve these problems at the cost of longer computation times (the time is proportional to ![]() ). This option is also useful for testing and improving convergence of atomic background calculations.
). This option is also useful for testing and improving convergence of atomic background calculations.
SCF rfms1 [lfms1 nscmt ca] (Useful)
This card controls FEFF's automated self-consistent potential calculations. Thus all fields except rfms1 are optional. If this card is not specified then all calculations are done with the non self-consistent (overlapped atomic) potential. By default lfms1=0, nscmt=30 and ca=0.2.
* Automated FMS SCF potentials for a molecule of radius 3.1 Angstroms SCF 3.1 1
INTERSTITIAL inters vtot (Advanced)
* improve interstitial density for ZnS structures. * vtot = (unit_cell_volume/number_of_atoms_in_unit_cell)/ratmin**3)=1.54 INTERSTITIAL 0 1.54
* Simulates effective ionization for formal valence state +1 * ipot, ionization ION 1 0.2
This card is used to specify the type of spin-dependent calculation. The complete description is given in Section G.1, when dealing with spin-dependent calculations.